|
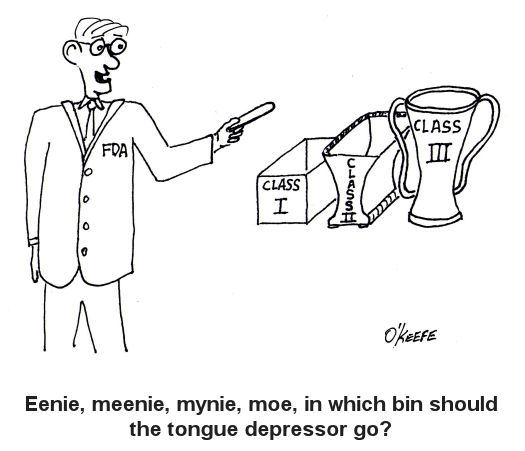
For the last couple of weeks we’ve been discussing FDA medical device risk classifications, namely Classes I, II, and III. We also began discussing the FDA system of regulatory controls governing each class, starting with General Controls. This week we’ll examine the more stringent guidelines that come into play within Special Controls. As you would imagine from the name, Special Controls come into play when General Controls aren’t deemed to be sufficient to deal with the situation. Class II and III medical devices, because they pose a higher level of risk to patients than Class I, generally require more FDA supervision than mere General Controls. These devices tend to fall under the auspices of Special Controls. Special Controls include things like special labeling requirements, complying with mandatory performance standards, and perhaps requiring that a manufacturer conduct a Post Market Surveillance (PMS) study. In case you’re wondering, a PMS may be required by the FDA to collect data after a medical device is sold, should there be any unexpected adverse events involving the device. A study of this data would aid in an investigation to determine the number of events, the cause of the events, and how to correct any problems that led to the events. Let’s look at some examples of how Special Controls apply to Class II medical devices. One example would be a cranial molding helmet. These helmets are often used with infants to reshape their skulls into becoming more symmetric. Due to the nature of this device’s application on such a delicate patient, Special Controls include a requirement for special labeling. In this case, the labeling must include warnings to physicians and parents that precautions must be taken during its application to protect patients from possible injury, including eye trauma and impairments of brain growth. Another example would be sutures. Yes, they are considered to be Class II medical devices. In this case, Special Controls require that sutures meet “mandatory performance standards.” What are “mandatory performance standards?” Well, they generally include industry consensus standards for particular medical devices. They are based on industry-wide accepted guidelines to ensure proper product performance. In this example, industry standards for suture material contain specific guidelines as to material composition, diameter size, mechanical strength, and biocompatibility. Adherence to these standards provides the highest assurance that sewn incisions won’t break open when the suture is stressed or the suture material won’t cause some sort of adverse reaction with the patient’s skin. As specific as Special Controls can be, they are sometimes not enough. On these occasions the FDA states, “Class III devices are those for which insufficient information exists to assure safety and effectiveness solely through General or Special Controls.” Under these circumstances more regulatory control may be imposed. This is the case when dealing with medical devices directly responsible for supporting/sustaining human life, such as a cardiac defibrillator. One such FDA control method that goes beyond Special Controls is the requirement to submit a Pre Market Approval application (PMA) to the FDA for approval. This PMA is subject to the most stringent FDA requirements. As a part of the PMA process a company must demonstrate the safety and effectiveness of a new medical device design by producing data and documentation obtained during “adequate and well-controlled” clinical trials. In our series on FDA Classifications for Medical Devices we have merely grazed the surface. Depending on the device in question there may be a myriad of other considerations, so please consult the FDA’s web site for the complete picture: http://www.fda.gov/MedicalDevices/default.htm. _____________________________________________ |

Tags: engineering expert witness, FDA, FDA Class I medical device, FDA Class II medical device, FDA Class III medical device, FDA general controls, FDA special controls, Food and Drug Administration, forensic engineer, medical device, medical device design, medical device risk, performance standards, PMA, PMS, post market surveylence, premarket approval application, special labeling




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}