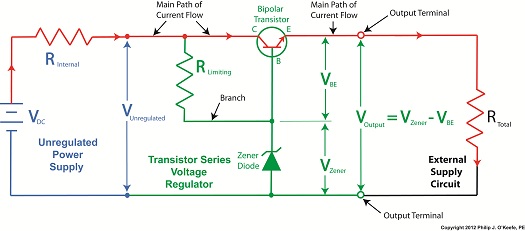
| Last time we introduced the first phase of the systems engineering approach to medical device design, the Concept Phase. It’s a phase which emphasizes clear communication between design engineers and stakeholders in the project. Effective communication results in clearly defined stakeholder requirements.
Stakeholder requirements are of three basic types, serving the needs of functionality, performance, and constraint. A functional requirement is a basic starting point, specifying the tasks to be accomplished by the system and its basic operation. For example, a medical diagnostic imaging device can be required to operate on a dedicated 15 amp branch circuit when plugged into a 115 volt wall outlet. Performance requirements delineate specific expectations as regards the system’s functionality, typically specifying parameters within which the system is to perform. For example, a medical device can be required to process a minimum number of test samples per hour. As for constraint requirements, they’re typically concerned with satisfactorily complying with both governmental regulations and industry standards. Governmental regulations include those established by the US Food and Drug Administration, as well as any applicable foreign agencies that may be involved should the medical device be introduced to other countries. Once the draft specification is written, design engineers review it in painstaking detail. Stakeholders’ requirements must all be accounted for and interpreted correctly. This is typically done by sitting down with each stakeholder and reviewing the draft. Finally, each stakeholder’s signature is obtained, signifying their approval. Now complete, the approved specification is used as a roadmap to guide project development through to the next and final step of the concept stage, devising alternate medical device concepts. Why discuss alternate concepts? Just like consumer comparison shopping for TVs and other goods, you’ve got to have a basis of comparison in order to make a sound judgment. This holds true for the medical device industry as well as private households. Next time we’ll proceed to the next stage of the design process, Development. We’ll see how alternate concepts are evaluated to find the best overall option, thus beginning the actual design process leading to manufacture. ____________________________________________
|
Posts Tagged ‘Food and Drug Administration’
Systems Engineering In Medical Device Design – Concept and Requirements
Monday, December 10th, 2012
Systems Engineering In Medical Device Design – Introduction
Monday, November 26th, 2012
Food Manufacturing Challenges – HACCP Design Principle No. 2
Sunday, October 23rd, 2011| What would you do if you heard an unfamiliar sound coming from your water heater? If you’re like most people you’d make a mental note to keep an eye on it, but ignore it for the most part. Unfortunately, this less than proactive approach often results in water heater floods. As an engineer, I’m more likely than the general population to investigate the cause of the water heater’s sound and proactively seek a remedy before a real problem has a chance to develop.
The FDA’s Hazard Analysis Critical Control Point (HACCP) seeks to accomplish the same with regard to food production. As discussed last week with regard to HACCP Principle 1, those involved in designing food processing equipment must proactively analyze designs to identify potential food safety hazards. Now let’s see how common sense is once again employed through Principle 2, guiding design engineers to take control of situations where hazards have been identified through Principle 1. HACCP Principle 2: Identify critical control points. A critical control point (CCP) is a step in the design process at which a control can be most effectively introduced to prevent or eliminate hazards. In this context a “control” would be a design revision to eliminate hazards identified during the Principle 1 stage. We will once again use the two examples introduced in last week’s blog discussion on Principle 1. In our first example, hazard analysis revealed that food can accumulate in a food processing machine in areas where cleaning is difficult or impossible. This accumulation would eventually rot and fall into uncontaminated food passing through production lines. Design engineers would work to address this contamination hazard by identifying a CCP within the design process, that is, the best place where a preventative measure can be added to the machine setup to facilitate removal of the accumulation. At that CCP, measures can be taken to change the machine’s design. Perhaps all that is needed to correct the situation is to include easy to remove access covers. In our second example hazard analysis revealed that the metal tooling as designed for our food production machine was too fragile and would not withstand the repeated forces imposed on it by the mass production process. This design flaw presents a strong possibility that metal parts will break off and enter food on the line. To correct this situation, design engineers must once again identify the juncture within the design process at which a CCP is identified. There, a preventative measure can most effectively be introduced, enabling more robust metal to be used in the tooling. The previous two examples illustrate CCPs being utilized within the design process. CCPs can also be introduced outside the design process, as when they are identified during the course of training procedures involving the operation, cleaning, and general maintenance of equipment and production lines. And an excellent way of implementing this approach is to have design engineers collaborate with operating and maintenance staff. Working together, they are best able to identify key elements to be addressed and make note of them within written procedures. Now that we have identified some examples of CCPs within the design process, we can move on to HACCP Principle 3 and how it guides design engineers to establish critical limits for each CCP.
|

Medical Device Design Controls – Transfer, Changes, and History
Sunday, September 12th, 2010| Did you ever hear the saying, “garbage in, garbage out?” Perhaps you’ve used it yourself at times, as when your teenager insists on writing their 20-page term paper the night before it’s due. Parents, having the benefit of decades of life experience, know that the outcome of a last ditch effort of this type will most likely not turn out well.
This wisdom also applies particularly well to the medical manufacturing process. The FDA is like the parent in this instance, mandating that Design Transfer Procedures be in place to avert the types of disasters which might ensue if the “garbage” philosophy were carried out. Meant to ensure that medical device designs are correctly translated into production specifications for manufacturing, Design Transfer Procedures keep those directly involved with the manufacturing process in check. It is absolutely vital that those involved in manufacturing receive accurate and complete information. Imagine what would happen if an engineer provided a manufacturer with faulty design information. Components could be made to the wrong specifications or of a material that proves toxic to the application. These errors range in negative effect from being costly in terms of dollars wasted to perhaps costing someone their life. A Design Transfer Procedure would ensure that a variety of mishaps do not occur during the transfer process. The procedure is typically overseen by the medical device company’s management. For example, a Design Transfer Procedure would lay out responsibilities of supervisors and managers to make sure the latest revision of electrical schematics, bills of materials, Gerber files, and quality testing procedures are received by the manufacturer of a device’s printed circuit boards. It’s important that the order is received in a timely manner so as not to hold up the manufacturing process. However, it’s much more important that the printed circuit board is made properly, the correct electrical components are placed on it in the correct orientation, and it is tested to make sure it doesn’t malfunction after assembly. Design Change Procedures basically ensure that when changes are necessary, the medical device company follows all the procedures for Design and Development Planning, Design Input, Design Output, Design Review, Design Verification, and Design Validation. Once the changes are reviewed, validated, verified, and approved, they can be incorporated into the original device design. This is where the Design Change Procedure must dovetail with the Design Transfer Procedure to make sure the correct information is provided to the company’s management staff in the procurement, manufacturing, product service, and warehouse departments. This is to make sure they can keep component vendors on track with the changes, maintain sufficient inventory of the changed components, put the right components in the device during assembly, and properly support repair technicians in the field. Yet another aspect of Design Controls promulgated by the FDA comes into play with the establishment of procedures for maintaining a Design History File (DHF). This DHF contains all documentation created during the life cycle of the project, meaning, movement from creation to completion and on into market introduction, sometimes beyond. DHF Procedure sets up protocols for collection and organization of information about the medical device design, starting with design documentation and covering the gamut from design changes, to validation testing, to design verification, and on to design review. All this is done to ensure that the initial product design was developed in accordance with the original design plan and overall product design requirements. _____________________________________________ |

Medical Device Design Controls – Output, Verification, Review, and Validation
Sunday, September 5th, 2010| Recently my wife was on a quest to make the perfect pound cake, but before she put butter to flour she did her research. What’s the best butter? Best flour? Eventually she came up with a recipe she felt would prove to be the Queen of all pound cakes. After the recipe came reviews by her test panel, or family members, including myself. Questions were asked, such as, When you first bite into a pound cake, do you want to be aware of vanilla or lemon? It was only then that she would begin to combine ingredients for the final mouth watering product. Very much this same procedure is used when coming up with a new medical device.
Previously we’ve discussed FDA requirements for medical devices as they concern design controls with respect to design and development planning and design input procedures. We’ll now focus on requirements for Design Output, Design Verification, and Design Review Procedures. Design Output and Design Verification Procedures go hand in hand to ensure that design output is properly documented, organized, reviewed, and evaluated in light of design input. What this means is that medical device companies must scrutinize and evaluate what is going into the design process, then make a comparison to what is coming out. The design is ultimately verified when all requirements for the medical device as previously set out have been met. “Design output” is just another name for work product after major phases of the design project are completed, such as when my wife determined which butter would produce the best pound cake. Design output typically takes the form of specifications, notes, calculations, computer programs, mechanical drawings, electrical schematics, printed circuit board (PCB) layouts, bills of materials (BOM), mockups, prototypes, test data, and test reports. These are then utilized by people outside engineering circles to manufacture components and assemble them into a final product. Design Review Procedures ensure that the design output is evaluated by others not directly involved in or responsible for the design work product, much as when family members served as a reviewing committee for my wife’s inquiries into taste preferences in pound cake. Sometimes she’d even ask a friend or neighbor to put their two cents in, and companies, too, will at times go outside and hire consultants to perform this function. By so doing, unbiased opinions are sought out, in the hopes that this fresh set of eyes will be more likely to spot errors, omissions, and misinterpretations that could prove disastrous if put into play. Design reviews are typically conducted after each major phase of a design project is complete. Just as a recipe that looks good on paper may not necessarily taste good, a device design will often seem to work perfectly on paper, then prove otherwise when its manufacture begins or it’s used in the field. Ideally bugs are worked out before the product hits production and, later, the marketplace. Design Validation Procedures make use of prototypes for testing and careful evaluation under simulated or actual use conditions. Does the design safely meet requirements for intended use? Does it conform with industry standards? If not, there’ll be a lot of wasted “dough” going into the trash — pun intended! Next time we’ll explore FDA requirements for Design Transfer and Design Changes. We’ll also talk about procedures for Design History Files. _____________________________________________ |

Medical Device Design Controls – Planning and Input
Sunday, August 29th, 2010| Have you ever had the divine experience of remodeling a major-use room of your house? Was the general contractor you employed able to understand what you wanted, plan out the work according to your requirements, and finish the job to your satisfaction? Maybe you had the unfortunate experience of hiring one that forgot your requirements, made things up as they went along, and stuck you with a room that looked awful, violated building codes, and didn’t meet your needs.
Now imagine what would happen if a medical device company took this haphazard approach to designing new products. Suppose the company’s engineers ignored the input of regulatory, marketing, procurement, quality control, and manufacturing staff? What if they chose not to follow applicable industry standards for performance and safety? And what if they failed to check design calculations or test prototypes for errors before putting the device into production and introducing it to the marketplace? The result is likely to be unfavorable, just like your contractor forgetting that you wanted a black granite countertop, not a beige one. To help eliminate painful and costly scenarios such as these, the FDA requires that medical device manufacturers establish and maintain procedures to control the design of Class II and III devices, and even some Class I. This requirement for a system of design controls is part of the Quality System Regulation (QSR) under Title 21 of the Code of Federal Regulations. In case you’re not too familiar with the Code of Federal Regulations, Title 21 gives the FDA legal authority to regulate food, drugs, and medical devices in the United States. So what falls under the premises of FDA design controls? Well, the FDA requires that a medical device company develop procedures for:
For now, let’s focus on Design and Development Planning and Design Input Procedures. In the Design and Development Planning Procedure companies must carefully plan who will be involved in each phase of product development, as well as how they will interact, all in an effort to ensure that information flows and design requirements are met. The right pool of people would include design engineers, in addition to those employees responsible for making sure that regulations are complied with and those who are charged with securing intellectual property rights to the design. Then there are those who must acquire the physical materials required to manufacture the device and those who will do the actual manufacturing of it. Also, those responsible for quality control, marketing, sales, and product service should be involved. Perhaps others should be involved as well. Mind you, the Design and Development Planning Procedures are not set in stone. They must be regularly reviewed and updated as the project evolves. Now let’s talk about Design Input, which is another term for a design requirement. These inputs can come from inside or outside the company. An example of a requirement coming from within is when Marketing stipulates that the maximum manufacturing cost of the device should not exceed $150 in order to maintain an acceptable margin of profit and be most competitive in the marketplace. A design requirement coming from outside the company would include industry standards that make specific requirements, such as requiring that the device in question be designed to protect its electronics from radio frequency interference. Next time we’ll continue our discussion on medical device design by exploring Design Output, Design Verification, and Design Review Procedures. _____________________________________________ |

FDA Classifications for Medical Devices – Special Controls
Monday, August 23rd, 2010|
For the last couple of weeks we’ve been discussing FDA medical device risk classifications, namely Classes I, II, and III. We also began discussing the FDA system of regulatory controls governing each class, starting with General Controls. This week we’ll examine the more stringent guidelines that come into play within Special Controls. As you would imagine from the name, Special Controls come into play when General Controls aren’t deemed to be sufficient to deal with the situation. Class II and III medical devices, because they pose a higher level of risk to patients than Class I, generally require more FDA supervision than mere General Controls. These devices tend to fall under the auspices of Special Controls. Special Controls include things like special labeling requirements, complying with mandatory performance standards, and perhaps requiring that a manufacturer conduct a Post Market Surveillance (PMS) study. In case you’re wondering, a PMS may be required by the FDA to collect data after a medical device is sold, should there be any unexpected adverse events involving the device. A study of this data would aid in an investigation to determine the number of events, the cause of the events, and how to correct any problems that led to the events. Let’s look at some examples of how Special Controls apply to Class II medical devices. One example would be a cranial molding helmet. These helmets are often used with infants to reshape their skulls into becoming more symmetric. Due to the nature of this device’s application on such a delicate patient, Special Controls include a requirement for special labeling. In this case, the labeling must include warnings to physicians and parents that precautions must be taken during its application to protect patients from possible injury, including eye trauma and impairments of brain growth. Another example would be sutures. Yes, they are considered to be Class II medical devices. In this case, Special Controls require that sutures meet “mandatory performance standards.” What are “mandatory performance standards?” Well, they generally include industry consensus standards for particular medical devices. They are based on industry-wide accepted guidelines to ensure proper product performance. In this example, industry standards for suture material contain specific guidelines as to material composition, diameter size, mechanical strength, and biocompatibility. Adherence to these standards provides the highest assurance that sewn incisions won’t break open when the suture is stressed or the suture material won’t cause some sort of adverse reaction with the patient’s skin. As specific as Special Controls can be, they are sometimes not enough. On these occasions the FDA states, “Class III devices are those for which insufficient information exists to assure safety and effectiveness solely through General or Special Controls.” Under these circumstances more regulatory control may be imposed. This is the case when dealing with medical devices directly responsible for supporting/sustaining human life, such as a cardiac defibrillator. One such FDA control method that goes beyond Special Controls is the requirement to submit a Pre Market Approval application (PMA) to the FDA for approval. This PMA is subject to the most stringent FDA requirements. As a part of the PMA process a company must demonstrate the safety and effectiveness of a new medical device design by producing data and documentation obtained during “adequate and well-controlled” clinical trials. In our series on FDA Classifications for Medical Devices we have merely grazed the surface. Depending on the device in question there may be a myriad of other considerations, so please consult the FDA’s web site for the complete picture: http://www.fda.gov/MedicalDevices/default.htm. _____________________________________________ |

FDA Classifications for Medical Devices – General Controls
Sunday, August 15th, 2010|
When I was a kid in Chicago back in the 1960s there was a show on television called Bozo’s Circus. Lucky kids were picked from the audience to play a bucket game. There were six buckets in a row, as I remember, each about a foot from the last. The kids had to stand in front of the first bucket to play. By the time the kids got to throw their ball into Bucket No. 6 there was probably ten feet for the ball to travel. So what would happen if the ball didn’t sink into the desired bucket, which would happen more often than not it seemed? Then Ringmaster Ned would direct Cookie the Clown to chase down the rogue balls. So what does this have to do with the FDA and medical devices? Well, in the loosest of terms you may think of the FDA’s classification system as Buckets 1 through 6 and Ringmaster Ned as the regulatory agent of the game. Okay, I’m really stretching on this analogy, but I did want to introduce some levity into the discussion! Last week we discussed the fact that the FDA classifies medical devices into three main categories, Classes I, II and III, Class I devices posing the least risk to patients, Class III the most. Now we’ll see how the FDA regulatory control system functions to oversee the medical devices within each classification. To begin with, you should be aware that regulatory controls are themselves divided into General and Special Control categories. In this article we’ll focus on General Controls. General Controls can apply to medical devices within all three FDA risk classifications. They include requirements for: — Registering of medical device manufacturers, distributors, repackagers, and relabelers with the FDA. This registration basically lets the FDA know they exist as an entity, and it gives the Agency information about who to contact should the need arise. — Listing medical devices with the FDA so the Agency can keep track of what kind of devices are being marketed in the United States. — Manufacturing devices in accordance with FDA Good Manufacturing Practices (GMP). GMP regulations require a quality approach to manufacturing, an approach which is designed to minimize or eliminate instances of contamination, defect, and error which could contribute to harm or kill a patient. GMP regulations address issues like sanitation, quality control, complaint handling, and record keeping. Effective complaint handling and record keeping systems are key in identifying and resolving issues that may pose increased risk to patients. — Labeling devices. The FDA requires that medical device labeling provide explicit directions for use. The labels must also contain appropriate warnings as needed to ensure the safe and effective use of the device. — Submitting a Premarket Notification to the FDA for approval before marketing a device, also referred to as a 510(k) within the industry. This name comes from the section of the federal regulation that deals with it, that is to say, companies must submit 510(k) documents to the FDA to demonstrate that the device they wish to market is comparably safe and effective as other equivalent devices already on the market. A 510(k) can only be submitted if Premarket Approval (PMA) for the device is not required. We’ll talk more about this in a future article on Special Controls. Now, because they pose such a low level of risk, many Class I medical devices are exempt from the requirement for 510(k) submissions altogether, and they may even be exempt from compliance with GMP regulations. In a nutshell, General Controls help the FDA keep track of what products are being sold by whom, and how effective and safe those products are. It also provides guidelines to medical device companies to help ensure their introduction of safe and effective products to the public. So what happens when a device falls outside of the parameters of the General Controls watch? That’s when more stringent guidelines come into play, for it has now entered the realm of Special Controls. _____________________________________________ |

FDA Classifications for Medical Devices
Sunday, August 8th, 2010|
Hardly a week goes by that the FDA, that is the Food and Drug Administration, is not in the news. From the recall of drugs found to be harmful after the fact, to investigations of medical device suppliers and inspections of salmonella contamination at meat processing plants, the FDA is responsible for overseeing and regulating a wide range of products and processes. It’s stated purpose being: The FDA is responsible for protecting the public health by assuring the safety, efficacy, and security of human and veterinary drugs, biological products, medical devices, our nation’s food supply, cosmetics, and products that emit radiation. http://www.fda.gov/AboutFDA/WhatWeDo/default.htm From its humble beginnings at the beginning of the last century as the Pure Food and Drug Act, it has grown to regulate more than $1 trillion worth of consumer goods, about 25% of that said to be attributable to consumer goods expenditures in the United States. In 1976, the FDA began classifying medical devices, using a three-tiered system to distinguish them according to level of risk to patients. Class I devices present the lowest level of risk and requires the least regulatory control, while Classes II and III represent higher levels of risk. Just to give you some perspective, Class I medical devices include things like tongue depressors, bedpans, arm slings, and hand-held surgical instruments. In the Class II category are things like surgical drapes, blood pressure cuffs, catheters, wheelchairs, heating pads, and x-ray film processing machines. And I’m sure you’ve guessed by now that Class III devices are for the heavy-hitters, including things like defibrillators, heart valves, and implanted cerebral stimulators. So why does the FDA classify medical devices? Practicality is one key reason. It would be highly impractical, if not downright impossible, for the FDA to subject manufacturers of low risk Class I devices, like tongue depressors, to the same scrutiny as manufacturers of Class III devices, such as heart valves, etc. Along with miles of red tape would come a huge financial burden that would effectively raise the price of your common tongue depressor through the roof and, undoubtedly, force the manufacturer out of business. Hand in hand with medical device classifications are regulatory controls imposed by the FDA. We’ll see how they fit into the picture next time. _____________________________________________ |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}