| When I was a kid I had a toy robot that captured my attention like no other toy. I thought it was so cool to have something animated that looked both humanoid and machine-like at the same time. It couldn’t do much, just walk in a stiff, jerky way and move its arms up and down, but that was enough to keep me fascinated.
Today’s generation of robots do not often take on the humanoid form, but they’re capable of so much more. Robots on assembly lines perform a variety of tasks like welding and placing electronic components on circuit boards, and they do it much more quickly and accurately than any human could, so they’re often employed in manufacturing. We’ve been discussing the Production stage of the systems engineering approach to medical device design. We learned that within the manufacturing process there are often opportunities for cost reduction, and today we’ll see how robots can be used to reach those goals. Last week we presented a sample scenario involving the manufacture of a percussion therapy device. In their quest to reduce manufacturing costs, engineers identified bottlenecks along the assembly line which led to idle worker time and the inability to keep up with orders. In addition to these production woes, it was discovered that the tedious, repetitive manual labor that occurred at each bottleneck created opportunities for assembly mistakes. As many as 30 devices per day were being rejected by quality control inspectors due to issues such as faulty wiring and improper parts usage. This led to expensive rework to correct mistakes. After further evaluation, design engineers determine that bottlenecks can be eliminated by installing automated assembly equipment in the three distinct assembly stages represented on the line, those involving wiring harnesses, printed circuit boards, and the motor drive mechanism. The potential for human error is high during many facets of manufacturing, and this can be minimized or eliminated through the use of robots, that is to say, mechanized equipment capable of automatically performing a complex series of specific tasks. These robots never tire of performing tedious, repetitive work, and their efficiency is unparalleled. Their introduction at key junctures on the assembly line has benefits across the manufacturing process, enabling workers to keep continuously busy and reducing the incidence of human error. The introduction of robotics is known as industrial automation. Robots efficiently increase manufacturing speed, and along with it profits, so their introduction more than compensates for the investment costs associated with purchasing them. Next time we’ll continue our look at the Production stage to discover another way that systems engineering can simplifying the assembly process, by eliminating some functions altogether. ___________________________________________
|
Posts Tagged ‘quality control’
Systems Engineering In Medical Device Design – Production, Part 3
Sunday, March 10th, 2013
Systems Engineering In Medical Device Design – Instructions, Part 3
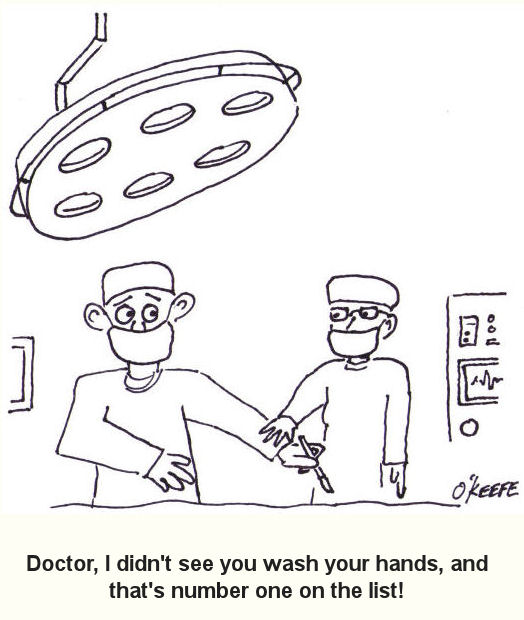
Sunday, January 20th, 2013| Last time we looked at the objective of the quality control department in the Production stage, that being to ensure that the end product produced fits all requirements. We learned that to meet this objective a great deal of collaboration must take place in the Development stage between quality control staff and design engineers in order to produce a complete set of instructions for quality control inspection and testing. Now let’s see how these instructions are developed.
Inspection and test methods are devised by the quality control department to ensure that a completed medical device lives up to its intended use. What good is a diagnostic imaging machine that doesn’t provide accurate internal views of a patient’s body? Or a heart defibrillator that sends electrical energy pulses to a patient’s heart muscles when it’s not supposed to? Quality control instructions are developed to guide inspection and testing methods so they’re performed correctly and consistently during the medical device Production stage. The objective is to catch problems before they occur. For example, it can be specified that the plastic body components of a medical device be visually inspected after they are received from an outside vendor to check for undesirable defects, such as the presence of burrs, cracks, or non-uniform coloration. If anomalies are discovered, they’re documented, and the components are rejected. In other words, they are barred from being used in the assembly process. Quality testing methods are varied. They may involve hooking up the completed medical device to test instruments to simulate all possible modes of operation and any anticipated glitches that may occur during testing. While hooked up, the device’s operation is measured against key parameters to ensure that all is working well, and the data gathered is recorded and analyzed to see if operation is within normal limits. For example, an electric multimeter could be connected to the power cord of the device to measure how much electrical current is being drawn from the wall outlet during operation. If current drawn is too high, it may be indicative of a defective electrical component, and an in depth examination would follow. Generally speaking, if test measurement parameters do not fall within acceptable limits as determined by previously established stakeholder requirements, then the medical device will be rejected by quality control. It will then be sent back to the manufacturing department, along with a detailed inspection and test report explaining why it was rejected. At this point the rejected device is either reworked or disposed of entirely. Next time we’ll continue our discussion on the development of instructions for the Utilization stage, the stage where the medical device is actually put into use by the end user. ___________________________________________
|

Systems Engineering In Medical Device Design – Instructions, Part I
Monday, January 7th, 2013| You know those instructional inserts that come in just about everything you buy? If you’re lucky they’re a one-pager, showing a simple illustration of how your purchase works. But sometimes they’re multiple pages long, even approaching the length of a short story. This is often the case when the item in question is complex and contains many parts.
If you’re like some people you try to avoid reading these instructions, preferring to forge ahead to the assembly/usage stage as quickly as possible, and you’ve probably had your fair share of times that this approach didn’t pan out. You were forced to re-do things and crack open the instruction manual anyway. If the instructions were written clearly, you may have eventually come to regard them as indispensable. Clearly written instructions are one of the desired end results of the Development stage of the systems engineering approach to medical device design that we’ve been discussing. These instructions flow naturally from the finalized detailed design which has been produced earlier in this stage. Instructions aid consumers in the assembly, usage, and maintenance of the device, making for a satisfied customer. Instructions also aid in the efficient and proper manufacture of devices. Without them assembly personnel wouldn’t work as efficiently, and the end result might not be a desirable one. It’s easy for parts to end up where they don’t belong, adjustments to be off, etc. Just think about the last “assemble it yourself at home” project you were involved in. The desired result is for instructions produced to be well defined and capable of instructing line assembly personnel in the actual construction of the medical device that takes place during the Production stage. Subjects such as parts identification, assembly procedures, and layout of assembly lines are discussed, all of which are needed to plan out the manufacturing process effectively. The objective is to manufacture the devices in a cost effective manner and with minimum probability of defects. Next week we’ll continue our discussion on instructions, focusing on those that are produced during the Development stage that serve the purpose of guiding quality control technicians during the Production stage. ___________________________________________
|

FDA Classifications for Medical Devices – General Controls
Sunday, August 15th, 2010|
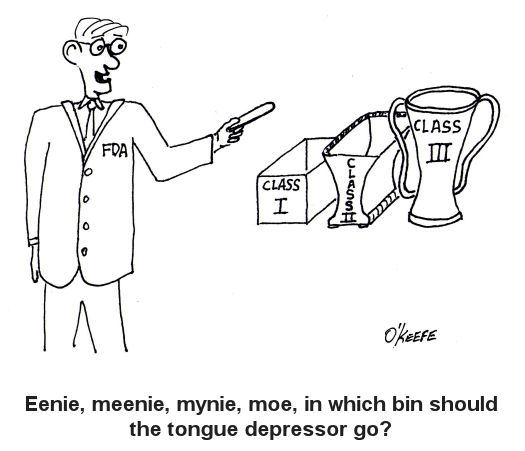
When I was a kid in Chicago back in the 1960s there was a show on television called Bozo’s Circus. Lucky kids were picked from the audience to play a bucket game. There were six buckets in a row, as I remember, each about a foot from the last. The kids had to stand in front of the first bucket to play. By the time the kids got to throw their ball into Bucket No. 6 there was probably ten feet for the ball to travel. So what would happen if the ball didn’t sink into the desired bucket, which would happen more often than not it seemed? Then Ringmaster Ned would direct Cookie the Clown to chase down the rogue balls. So what does this have to do with the FDA and medical devices? Well, in the loosest of terms you may think of the FDA’s classification system as Buckets 1 through 6 and Ringmaster Ned as the regulatory agent of the game. Okay, I’m really stretching on this analogy, but I did want to introduce some levity into the discussion! Last week we discussed the fact that the FDA classifies medical devices into three main categories, Classes I, II and III, Class I devices posing the least risk to patients, Class III the most. Now we’ll see how the FDA regulatory control system functions to oversee the medical devices within each classification. To begin with, you should be aware that regulatory controls are themselves divided into General and Special Control categories. In this article we’ll focus on General Controls. General Controls can apply to medical devices within all three FDA risk classifications. They include requirements for: — Registering of medical device manufacturers, distributors, repackagers, and relabelers with the FDA. This registration basically lets the FDA know they exist as an entity, and it gives the Agency information about who to contact should the need arise. — Listing medical devices with the FDA so the Agency can keep track of what kind of devices are being marketed in the United States. — Manufacturing devices in accordance with FDA Good Manufacturing Practices (GMP). GMP regulations require a quality approach to manufacturing, an approach which is designed to minimize or eliminate instances of contamination, defect, and error which could contribute to harm or kill a patient. GMP regulations address issues like sanitation, quality control, complaint handling, and record keeping. Effective complaint handling and record keeping systems are key in identifying and resolving issues that may pose increased risk to patients. — Labeling devices. The FDA requires that medical device labeling provide explicit directions for use. The labels must also contain appropriate warnings as needed to ensure the safe and effective use of the device. — Submitting a Premarket Notification to the FDA for approval before marketing a device, also referred to as a 510(k) within the industry. This name comes from the section of the federal regulation that deals with it, that is to say, companies must submit 510(k) documents to the FDA to demonstrate that the device they wish to market is comparably safe and effective as other equivalent devices already on the market. A 510(k) can only be submitted if Premarket Approval (PMA) for the device is not required. We’ll talk more about this in a future article on Special Controls. Now, because they pose such a low level of risk, many Class I medical devices are exempt from the requirement for 510(k) submissions altogether, and they may even be exempt from compliance with GMP regulations. In a nutshell, General Controls help the FDA keep track of what products are being sold by whom, and how effective and safe those products are. It also provides guidelines to medical device companies to help ensure their introduction of safe and effective products to the public. So what happens when a device falls outside of the parameters of the General Controls watch? That’s when more stringent guidelines come into play, for it has now entered the realm of Special Controls. _____________________________________________ |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}